
All Information was copied form Wikipedia articles for Transcription (Genetics), Translation (Genetics), Process of Reverse Transcription, Posttranslational modification, Primary Structure, DNA Replication, Epigenetics, and DNA Methylation. From the wikipedia article on the “Central Dogma Of Molecular Biology”
Central dogma of molecular biology
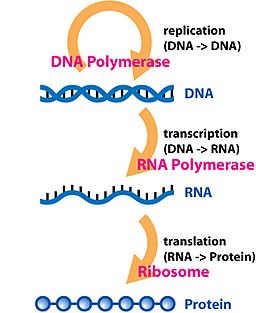
The central dogma of molecular biology describes the way genetic information is expected to be transferred in a single direction through a biological system. It was first stated by Francis Crick in 1958[1] and re-stated in a Nature paper published in 1970:[2]

Information flow in biological systems
- The central dogma of molecular biology deals with the detailed residue-by-residue transfer of sequential information. It states that such information cannot be transferred back from protein to either protein or nucleic acid.
Or, as Marshall Nirenberg said, “DNA makes RNA makes protein.”[3]
To appreciate the significance of the concept, note that Crick had misapplied the term “dogma” in ignorance. In evolutionary or molecular biological theory, either then or subsequently, Crick’s proposal had nothing to do with the correct meaning of “dogma”. He subsequently documented this error in his autobiography.
The dogma is a framework for understanding the transfer of sequence information between sequential information-carrying biopolymers, in the most common or general case, in living organisms. There are 3 major classes of such biopolymers: DNA and RNA (both nucleic acids), and protein. There are 3×3 = 9 conceivable direct transfers of information that can occur between these. The dogma classes these into 3 groups of 3: 3 general transfers (believed to occur normally in most cells), 3 special transfers (known to occur, but only under specific conditions in case of some viruses or in a laboratory), and 3 unknown transfers (believed never to occur). The general transfers describe the normal flow of biological information: DNA can be copied to DNA (DNA replication), DNA information can be copied into mRNA (transcription), and proteins can be synthesized using the information in mRNA as a template (translation).[2]
Biological sequence information
Primary structure
In biochemistry, the Primary structure of a biological molecule is the exact specification of its atomic composition and the chemical bonds connecting those atoms (including stereochemistry). For a typical unbranched, un-crosslinked biopolymer (such as a molecule of DNA, RNA or typical intracellular protein), the primary structure is equivalent to specifying the sequence of its monomeric subunits, e.g., thenucleotide or peptide sequence.
Primary structure is sometimes mistakenly termed primary sequence, but there is no such term, as well as no parallel concept of secondary or tertiary sequence. By convention, the primary structure of a protein is reported starting from the amino-terminal (N) end to the carboxyl-terminal (C) end, while the primary structure of DNA or RNA molecule is reported from the 5′ end to the 3′ end.
The primary structure of a nucleic acid molecule refers to the exact sequence of nucleotides that comprise the whole molecule. Frequently the primary structure encodes motifs that are of functional importance. Some examples of sequence motifs are: the C/D[1] and H/ACA boxes[2] of snoRNAs, Sm binding site found in spliceosomal RNAs such as U1, U2, U4, U5, U6, U12 and U3, the Shine-Dalgarno sequence,[3] the Kozak consensus sequence[4] and the RNA polymerase III terminator.[5]
The biopolymers that comprise DNA, RNA and amino acids are linear polymers (i.e.: each monomer is connected to at most two other monomers). The sequence of their monomers effectively encodes information. The transfers of information described by the central dogma are faithful, deterministic transfers, wherein one biopolymer’s sequence is used as a template for the construction of another biopolymer with a sequence that is entirely dependent on the original biopolymer’s sequence.
General transfers of biological sequential information

Transcription
Transcription (genetics)
Transcription is the process of creating a complementary RNA copy of a sequence of DNA.[1] Both RNA and DNA are nucleic acids, which use base pairs of nucleotides as a complementary language that can be converted back and forth from DNA to RNA by the action of the correct enzymes. During transcription, a DNA sequence is read by an RNA polymerase, which produces a complementary, antiparallel RNA strand. As opposed to DNA replication, transcription results in an RNA complement that includes uracil (U) in all instances where thymine (T) would have occurred in a DNA complement. Also unlike DNA replication where DNA is synthesised, transcription does not involve an RNA primer to initiate RNA synthesis.
Transcription is explained easily in 4 or 5 steps, each moving like a wave along the DNA.
- RNA polymerase moves the transcription bubble, a stretch of unpaired nucleotides, by breaking the hydrogen bonds between complementary nucleotides.
- RNA polymerase adds matching RNA nucleotides that are paired with complementary DNA bases.
- RNA sugar-phosphate backbone forms with assistance from RNA polymerase.
- Hydrogen bonds of the untwisted RNA + DNA helix break, freeing the newly synthesized RNA strand.
- If the cell has a nucleus, the RNA is further processed (addition of a 3′ poly-A tail and a 5′ cap) and exits through to the cytoplasm through the nuclear pore complex.
Transcription is the first step leading to gene expression. The stretch of DNA transcribed into an RNA molecule is called a transcription unit and encodes at least one gene. If the gene transcribed encodes aprotein, the result of transcription is messenger RNA (mRNA), which will then be used to create that protein via the process of translation. Alternatively, the transcribed gene may encode for either non-coding RNA genes (such as microRNA, lincRNA, etc.) or ribosomal RNA (rRNA) or transfer RNA (tRNA), other components of the protein-assembly process, or other ribozymes.[2]
A DNA transcription unit encoding for a protein contains not only the sequence that will eventually be directly translated into the protein (the coding sequence) but also regulatory sequences that direct and regulate the synthesis of that protein. The regulatory sequence before (upstream from) the coding sequence is called the five prime untranslated region (5’UTR), and the sequence following (downstream from) the coding sequence is called the three prime untranslated region (3’UTR).[2]
Transcription has some proofreading mechanisms, but they are fewer and less effective than the controls for copying DNA; therefore, transcription has a lower copying fidelity than DNA replication.[3]
As in DNA replication, DNA is read from 3′ → 5′ during transcription. Meanwhile, the complementary RNA is created from the 5′ → 3′ direction. This means its 5′ end is created first in base pairing. Although DNA is arranged as two antiparallel strands in a double helix, only one of the two DNA strands, called the template strand, is used for transcription. This is because RNA is only single-stranded, as opposed to double-stranded DNA. The other DNA strand is called the coding (lagging) strand, because its sequence is the same as the newly created RNA transcript (except for the substitution of uracil for thymine). The use of only the 3′ → 5′ strand eliminates the need for the Okazaki fragments seen in DNA replication.[2]
Transcription is divided into 5 stages: pre-initiation, initiation, promoter clearance, elongation and termination.[2]
Major steps
Stage I: Pre-initiation
In eukaryotes, RNA polymerase, and therefore the initiation of transcription, requires the presence of a core promoter sequence in the DNA. Promoters are regions of DNA that promote transcription and, in eukaryotes, are found at -30, -75, and -90 base pairs upstream from the transcription start site (abbreviated to TSS). Core promoters are sequences within the promoter that are essential for transcription initiation. RNA polymerase is able to bind to core promoters in the presence of various specific transcription factors.[citation needed]
The most characterized type of core promoter in eukaryotes is a short DNA sequence known as a TATA box, found 25-30 base pairs upstream from the TSS.[citation needed] The TATA box, as a core promoter, is the binding site for a transcription factor known as TATA-binding protein (TBP), which is itself a subunit of another transcription factor, called Transcription Factor II D (TFIID). After TFIID binds to the TATA box via the TBP, five more transcription factors and RNA polymerase combine around the TATA box in a series of stages to form a preinitiation complex. One transcription factor, Transcription factor II H, has two components with helicase activity and so is involved in the separating of opposing strands of double-stranded DNA to form the initial transcription bubble. However, only a low, or basal, rate of transcription is driven by the preinitiation complex alone. Other proteins known as activators and repressors, along with any associated coactivators or corepressors, are responsible for modulating transcription rate.[citation needed]
Thus, preinitiation complex contains:[citation needed] 1. Core Promoter Sequence 2. Transcription Factors 3. RNA Polymerase 4. Activators and Repressors. The transcription preinitiation in archaea is, in essence, homologous to that of eukaryotes, but is much less complex.[4] The archaeal preinitiation complex assembles at a TATA-box binding site; however, in archaea, this complex is composed of only RNA polymerase II, TBP, and TFB (the archaeal homologue of eukaryotic transcription factor II B (TFIIB)).[5][6]
Stage II: Initiation
In bacteria, transcription begins with the binding of RNA polymerase to the promoter in DNA. RNA polymerase is a core enzymeconsisting of five subunits: 2 α subunits, 1 β subunit, 1 β’ subunit, and 1 ω subunit. At the start of initiation, the core enzyme is associated with a sigma factor that aids in finding the appropriate -35 and -10 base pairs downstream of promoter sequences.[7] When the sigma factor and RNA polymerase combine, they form a holoenzyme.
Transcription initiation is more complex in eukaryotes. Eukaryotic RNA polymerase does not directly recognize the core promoter sequences. Instead, a collection of proteins called transcription factors mediate the binding of RNA polymerase and the initiation of transcription. Only after certain transcription factors are attached to the promoter does the RNA polymerase bind to it. The completed assembly of transcription factors and RNA polymerase bind to the promoter, forming a transcription initiation complex. Transcription in the archaea domain is similar to transcription in eukaryotes.[8]
Stage III: Promoter clearance
After the first bond is synthesized, the RNA polymerase must clear the promoter. During this time there is a tendency to release the RNA transcript and produce truncated transcripts. This is called abortive initiation and is common for both eukaryotes and prokaryotes.[9] Abortive initiation continues to occur until the σ factor rearranges, resulting in the transcription elongation complex (which gives a 35 bp moving footprint). The σ factor is released before 80 nucleotides of mRNA are synthesized.[10] Once the transcript reaches approximately 23 nucleotides, it no longer slips and elongation can occur. This, like most of the remainder of transcription, is an energy-dependent process, consuming adenosine triphosphate (ATP).[citation needed]
Promoter clearance coincides with phosphorylation of serine 5 on the carboxy terminal domain of RNAP II in eukaryotes, which is phosphorylated by TFIIH.[citation needed]
Stage IV: Elongation
One strand of the DNA, the template strand (or noncoding strand), is used as a template for RNA synthesis. As transcription proceeds, RNA polymerase traverses the template strand and uses base pairing complementarity with the DNA template to create an RNA copy. Although RNA polymerase traverses the template strand from 3′ → 5′, the coding (non-template) strand and newly-formed RNA can also be used as reference points, so transcription can be described as occurring 5′ → 3′. This produces an RNA molecule from 5′ → 3′, an exact copy of the coding strand (except that thymines are replaced with uracils, and the nucleotides are composed of a ribose (5-carbon) sugar where DNA has deoxyribose (one less oxygen atom) in its sugar-phosphate backbone).[citation needed]
Unlike DNA replication, mRNA transcription can involve multiple RNA polymerases on a single DNA template and multiple rounds of transcription (amplification of particular mRNA), so many mRNA molecules can be rapidly produced from a single copy of a gene.[citation needed]
Elongation also involves a proofreading mechanism that can replace incorrectly incorporated bases. In eukaryotes, this may correspond with short pauses during transcription that allow appropriate RNA editing factors to bind. These pauses may be intrinsic to the RNA polymerase or due to chromatin structure.[citation needed]
Stage V: Termination
Bacteria use two different strategies for transcription termination.1.Rho-independent transcription 2.Rho-dependent transcription. In Rho-independent transcription termination,also called intrinsic termination, RNA transcription stops when the newly synthesized RNA molecule forms a G-C-rich hairpin loop followed by a run of Us. When the hairpin forms, the mechanical stress breaks the weak rU-dA bonds, now filling the DNA-RNA hybrid. This pulls the poly-U transcript out of the active site of the RNA polymerase, in effect, terminating transcription. In the “Rho-dependent” type of termination, a protein factor called “Rho” destabilizes the interaction between the template and the mRNA, thus releasing the newly synthesized mRNA from the elongation complex.[11]
Transcription termination in eukaryotes is less understood but involves cleavage of the new transcript followed by template-independent addition of As at its new 3′ end, in a process called polyadenylation.[12]
Transcription is the process by which the information contained in a section of DNA is transferred to a newly assembled piece of messenger RNA (mRNA). It is facilitated by RNA polymerase and transcription factors. In eukaryotic cells the primary transcript (pre-mRNA) must be processed further in order to ensure translation. This normally includes a 5′ cap, a poly-A tail and splicing. Alternative splicing can also occur, which contributes to the diversity of proteins any single mRNA can produce.
Translation (biology and/or genetics)
In molecular biology and genetics, translation is the third stage of protein biosynthesis (part of the overall process of gene expression). In translation, messenger RNA (mRNA) produced by transcription is decoded by the ribosome to produce a specific amino acid chain, or polypeptide, that will later fold into an active protein. In Bacteria, translation occurs in the cell’s cytoplasm, where the large and small subunits of the ribosome are located, and bind to the mRNA. In Eukaryotes, translation occurs across the membrane of the endoplasmic reticulum in a process called vectorial synthesis. The ribosome facilitates decoding by inducing the binding of tRNAswith complementary anticodon sequences to that of the mRNA. The tRNAs carry specific amino acids that are chained together into a polypeptide as the mRNA passes through and is “read” by the ribosome in a fashion reminiscent to that of a stock ticker and ticker tape.
In many instances, the entire ribosome/mRNA complex bind to the outer membrane of the rough endoplasmic reticulum and release the nascent protein polypeptide inside for later vesicle transport and secretion outside of the cell. Many types of transcribed RNA, such as transfer RNA, ribosomal RNA, and small nuclear RNA, do not undergo translation into proteins.
Translation proceeds in four phases: initiation, elongation, translocation and termination (all describing the growth of the amino acid chain, or polypeptide that is the product of translation). Amino acids are brought to ribosomes and assembled into proteins.
In activation, the correct amino acid is covalently bonded to the correct transfer RNA (tRNA). The amino acid is joined by its carboxyl group to the 3′ OH of the tRNA by an ester bond. When the tRNA has an amino acid linked to it, it is termed “charged”. Initiation involves the small subunit of the ribosome binding to the 5′ end of mRNA with the help of initiation factors (IF). Termination of the polypeptide happens when the A site of the ribosome faces a stop codon (UAA, UAG, or UGA). No tRNA can recognize or bind to this codon. Instead, the stop codon induces the binding of arelease factor protein that prompts the disassembly of the entire ribosome/mRNA complex.
A number of antibiotics act by inhibiting translation; these include anisomycin, cycloheximide, chloramphenicol, tetracycline, streptomycin, erythromycin, and puromycin, among others. Prokaryotic ribosomes have a different structure from that of eukaryotic ribosomes, and thus antibiotics can specifically target bacterial infections without any detriment to a eukaryotic host’s cells.
Basic mechanisms
![]()
A ribosome translating a protein that is secreted into the endoplasmic reticulum. tRNAs are colored dark blue.

Tertiary structure of tRNA. CCA tail in orange, Acceptor stem in purple, D arm in red, Anticodon arm in blue with Anticodon in black, T arm in green.
The basic process of protein production is addition of one amino acid at a time to the end of a protein. This operation is performed by a ribosome. The choice of amino acid type to add is determined by an mRNA molecule. Each amino acid added is matched to a three nucleotide subsequence of the mRNA. For each such triplet possible, only one particular amino acid type is accepted. The successive amino acids added to the chain are matched to successive nucleotide triplets in the mRNA. In this way the sequence of nucleotides in the template mRNA chain determines the sequence of amino acids in the generated amino acid chain.[1]Addition of an amino acid occurs at the C-terminus of the peptide and thus translation is said to be amino-to-carboxyl directed.[2]
The mRNA carries genetic information encoded as a ribonucleotide sequence from the chromosomes to the ribosomes. The ribonucleotides are “read” by translational machinery in a sequence of nucleotide triplets called codons. Each of those triplets codes for a specific amino acid.
The ribosome molecules translate this code to a specific sequence of amino acids. The ribosome is a multisubunit structure containing rRNA and proteins. It is the “factory” where amino acids are assembled into proteins. tRNAs are small noncoding RNA chains (74-93 nucleotides) that transport amino acids to the ribosome. tRNAs have a site for amino acid attachment, and a site called an anticodon. The anticodon is an RNA triplet complementary to the mRNA triplet that codes for their cargo amino acid.
Aminoacyl tRNA synthetase (an enzyme) catalyzes the bonding between specific tRNAs and the amino acids that their anticodon sequences call for. The product of this reaction is an aminoacyl-tRNA molecule. This aminoacyl-tRNA travels inside the ribosome, where mRNA codons are matched through complementary base pairing to specific tRNA anticodons. The ribosome has three sites for tRNA to bind. They are the aminoacyl site (abbreviated A), the peptidyl site (abbreviated P) and the exit site (abbreviated E). With respect to the mRNA, the three sites are oriented 5’to 3’ E-P-A, because ribosomes moves toward the 3′ end of mRNA. The A site binds the incoming tRNA with the complementary codon on the mRNA. The P site holds the tRNA with the growing polypeptide chain. The E site holds the tRNA without its amino acid. When an aminoacyl-tRNA initially binds to its corresponding codon on the mRNA, it is in the A site. Then, a peptide bond forms between the amino acid of the tRNA in the A site and the amino acid of the charged tRNA in the P site. The growing polypeptide chain is transferred to the tRNA in the A site. Translocation occurs, moving the tRNA in the P site, now without an amino acid, to the E site; the tRNA that was in the A site, now charged with the polypeptide chain, is moved to the P site. The tRNA in the E site leaves and another aminoacyl-tRNA enters the A site to repeat the process.[3]
After the new amino acid is added to the chain, the energy provided by the hydrolysis of a GTP bound to the translocase EF-G (in prokaryotes) and eEF-2 (ineukaryotes) moves the ribosome down one codon towards the 3′ end. The energy required for translation of proteins is significant. For a protein containing n amino acids, the number of high-energy Phosphate bonds required to translate it is 4n-1[citation needed]. The rate of translation varies; it is significantly higher in prokaryotic cells (up to 17-21 amino acid residues per second) than in eukaryotic cells (up to 6-9 amino acid residues per second).[4]
Eventually, this mature mRNA finds its way to a ribosome, where it is translated. In prokaryotic cells, which have no nuclear compartment, the process of transcription and translation may be linked together. In eukaryotic cells, the site of transcription (the cell nucleus) is usually separated from the site of translation (the cytoplasm), so the mRNA must be transported out of the nucleus into the cytoplasm, where it can be bound by ribosomes. The mRNA is read by the ribosome as triplet codons, usually beginning with an AUG (adenine−uracil−guanine), or initiator methionine codon downstream of the ribosome binding site. Complexes of initiation factors and elongation factors bring aminoacylated transfer RNAs (tRNAs) into the ribosome-mRNA complex, matching the codon in the mRNA to the anti-codon on the tRNA, thereby adding the correct amino acid in the sequence encoding the gene. As the amino acids are linked into the growing peptide chain, they begin folding into the correct conformation. Translation ends with a UAA, UGA, or UAG stop codon. The nascent polypeptide chain is then released from the ribosome as a mature protein. In some cases the new polypeptide chain requires additional processing to make a mature protein. The correct folding process is quite complex and may require other proteins, called chaperone proteins. Occasionally, proteins themselves can be further spliced; when this happens, the inside “discarded” section is known as an intein.
DNA replication
DNA replication is a biological process that occurs in all living organisms and copies their DNA; it is the basis for biological inheritance. The process starts when one double-stranded DNA molecule produces two identical copies of the molecule. The cell cycle (mitosis) also pertains to the DNA replication/reproduction process. The cell cycle includes interphase, prophase, metaphase, anaphase, and telophase. Each strand of the original double-stranded DNA molecule serves as template for the production of the complementary strand, a process referred to as semiconservative replication. Cellular proofreading and error toe-checking mechanisms ensure near perfect fidelity for DNA replication.[1][2]
In a cell, DNA replication begins at specific locations in the genome, called “origins”.[3] Unwinding of DNA at the origin, and synthesis of new strands, forms a replication fork. In addition to DNA polymerase, the enzyme that synthesizes the new DNA by adding nucleotides matched to the template strand, a number of other proteins are associated with the fork and assist in the initiation and continuation of DNA synthesis.
DNA replication can also be performed in vitro (artificially, outside a cell). DNA polymerases, isolated from cells, and artificial DNA primers are used to initiate DNA synthesis at known sequences in a template molecule. The polymerase chain reaction (PCR), a common laboratory technique, employs such artificial synthesis in a cyclic manner to amplify a specific target DNA fragment from a pool of DNA.
DNA structure
DNA usually exists as a double-stranded structure, with both strands coiled together to form the characteristic double-helix. Each single strand of DNA is a chain of four types of nucleotides having the bases:adenine, cytosine, guanine, and thymine (commonly noted as A,C, G & T). A nucleotide is a mono-, di-, or triphosphate deoxyribonucleoside; that is, a deoxyribose sugar is attached to one, two, or three phosphates, and a base. Chemical interaction of these nucleotides forms phosphodiester linkages, creating the phosphate-deoxyribose backbone of the DNA double helix with the bases pointing inward. Nucleotides (bases) are matched between strands through hydrogen bonds to form base pairs. Adenine pairs with thymine (two hydrogen bonds), and cytosine pairs with guanine (three hydrogen bonds) because a purine must pair with a pyrimidine: a pyrimidine cannot pair with another pyrimidine because the strands would be very close to each other; in a purine pair, the strands would be too far apart and the structure would be unstable. If A-C paired, there would be one hydrogen not bound to anything, making the DNA unstable.
DNA strands have a directionality, and the different ends of a single strand are called the “3′ (three-prime) end” and the “5′ (five-prime) end” with the direction of the naming going 5 prime to the 3 prime region. The strands of the helix are anti-parallel with one being 5 prime to 3 then the opposite strand 3 prime to 5. These terms refer to the carbon atom in deoxyribose to which the next phosphate in the chain attaches. Directionality has consequences in DNA synthesis, because DNA polymerase can synthesize DNA in only one direction by adding nucleotides to the 3′ end of a DNA strand.
The pairing of bases in DNA through hydrogen bonding means that the information contained within each strand is redundant. The nucleotides on a single strand can be used to reconstruct nucleotides on a newly synthesized partner strand.[4]
DNA polymerase

DNA polymerases adds nucleotides to the 3′ end of a strand of DNA.[5] If a mismatch is accidentally incorporated, the polymerase is inhibited from further extension. Proofreading removes the mismatched nucleotide and extension continues.
DNA polymerases are a family of enzymes that carry out all forms of DNA replication.[6] However, a DNA polymerase can only extend an existing DNA strand paired with a template strand; it cannot begin the synthesis of a new strand. To begin synthesis, a short fragment of DNA or RNA, called a primer, must be created and paired with the template DNA strand.
DNA polymerase then synthesizes a new strand of DNA by extending the 3′ end of an existing nucleotide chain, adding new nucleotides matched to the template strand one at a time via the creation of phosphodiester bonds. The energy for this process of DNA polymerization comes from two of the three total phosphates attached to each unincorporated base. (Free bases with their attached phosphate groups are called nucleoside triphosphates.) When a nucleotide is being added to a growing DNA strand, two of the phosphates are removed and the energy produced creates a phosphodiester bond that attaches the remaining phosphate to the growing chain. The energetics of this process also help explain the directionality of synthesis – if DNA were synthesized in the 3′ to 5′ direction, the energy for the process would come from the 5′ end of the growing strand rather than from free nucleotides.
In general, DNA polymerases are extremely accurate, making less than one mistake for every 107 nucleotides added.[7] Even so, some DNA polymerases also have proofreading ability; they can remove nucleotides from the end of a strand in order to correct mismatched bases. If the 5′ nucleotide needs to be removed during proofreading, the triphosphate end is lost. Hence, the energy source that usually provides energy to add a new nucleotide is also lost.
Replication process
DNA Replication, like all biological polymerization processes, proceeds in three enzymatically catalyzed and coordinated steps: initiation, elongation and termination.
Origins
For a cell to divide, it must first replicate its DNA.[8] This process is initiated at particular points in the DNA, known as “origins”, which are targeted by proteins that separate the two strands and initiate DNA synthesis.[3] Origins contain DNA sequences recognized by replication initiator proteins (e.g., DnaA in E. coli’ and the Origin Recognition Complex in yeast).[9] These initiators recruit other proteins to separate the strands and initiate replication forks.
Initiator proteins recruit other proteins and form the pre-replication complex, which separate the DNA strands at the origin and forms a bubble. Origins tend to be “AT-rich” (rich in adenine and thymine bases) to assist this process, because A-T base pairs have two hydrogen bonds (rather than the three formed in a C-G pair)—in general, strands rich in these nucleotides are easier to separate since less energy is required to break relatively fewer hydrogen bonds.[10]
All known DNA replication systems require a free 3′ OH group before synthesis can be initiated (Important note: DNA is read in 3′ to 5′ direction whereas a new strand is synthesised in the 5′ to 3′ direction – this is entirely logical but is often confused). Four distinct mechanisms for synthesis have been described.
1. All cellular life forms and many DNA viruses, phages and plasmids use a primase to synthesize a short RNA primer with a free 3′ OH group which is subsequently elongated by a DNA polymerase.
2. The retroelements (including retroviruses) employ a transfer RNA that primes DNA replication by providing a free 3′ OH that is used for elongation by thereverse transcriptase.
3. In the adenoviruses and the φ29 family of bacteriophages, the 3′ OH group is provided by the side chain of an amino acid of the genome attached protein (the terminal protein) to which nucleotides are added by the DNA polymerase to form a new strand.
4. In the single stranded DNA viruses – a group that includes the circoviruses, the geminiviruses, the parvoviruses and others – and also the many phages and plasmids that use the rolling circle replication (RCR) mechanism, the RCR endonuclease creates a nick the genome strand (single stranded viruses) or one of the DNA strands (plasmids). The 5′ end of the nicked strand is transferred to a tyrosine residue on the nuclease and the free 3′ OH group is then used by the DNA polymerase for new strand synthesis.
The best known of these mechanisms is that used by the cellular organisms. In these once the two strands are separated, RNA primers are created on the template strands. To be more specific, the leading strand receives one RNA primer per active origin of replication while the lagging strand receives several; these several fragments of RNA primers found on the lagging strand of DNA are called Okazaki fragments, named after their discoverer. DNA polymerase extends the leading strand in one continuous motion and the lagging strand in a discontinuous motion (due to the Okazaki fragments). RNase removes the RNA fragments used to initiate replication by DNA polymerase, and another DNA Polymerase enters to fill the gaps. When this is complete, a single nick on the leading strand and several nicks on the lagging strand can be found. Ligase works to fill these nicks in, thus completing the newly replicated DNA molecule.
The primase used in this process differs significantly between bacteria and archaea/eukaryotes. Bacteria use a primase belonging to the DnaG protein superfamily which contains a catalytic domain of the TOPRIM fold type. The TOPRIM fold contains an α/β core with four conserved strands in a Rossmann-like topology. This structure is also found in the catalytic domains of topoisomerase Ia, topoisomerase II, the OLD-family nucleases and DNA repair proteins related to the RecR protein.
The primase used by archaea and eukaryotes in contrast contains a highly derived version of the RNA recognition motif (RRM). This primase is structurally similar to many viral RNA dependent RNA polymerases, reverse transcriptases, cyclic nucleotide generating cyclases and DNA polymerases of the A/B/Y families that are involved in DNA replication and repair. All these proteins share a catalytic mechanism of di-metal-ion-mediated nucleotide transfer, whereby two acidic residues located at the end of the first strand and between the second and third strands of the RRM-like unit respectively, chelate two divalent cations.
As DNA synthesis continues, the original DNA strands continue to unwind on each side of the bubble, forming a replication fork with two prongs. In bacteria, which have a single origin of replication on their circular chromosome, this process eventually creates a “theta structure” (resembling the Greek letter theta: θ). In contrast, eukaryotes have longer linear chromosomes and initiate replication at multiple origins within these.[citation needed]
As the final step in the central dogma, DNA replication must occur in order to faithfully transmit genetic material to the progeny of any cell or organism. Replication is carried out by a complex group of proteins called the replisome which consists of a helicase that unwinds the superhelix as well as the double-stranded DNA helix, and DNA polymerase and its associated proteins, which insert new nucleic in a sequence specific manner. This process typically takes place during S phase of the cell cycle.
Special transfers of biological sequential information
Reverse transcription

Process of reverse transcription
Reverse transcriptase creates single-stranded DNA from an RNA template.
In virus species with reverse transcriptase lacking DNA-dependent DNA polymerase activity, creation of double-stranded DNA can possibly be done by host-encoded DNA polymerase δ, mistaking the viral DNA-RNA for a primer and synthesizing a double-stranded DNA by similar mechanism as in primer removal, where the newly synthesized DNA displaces the original RNA template.
The process of reverse transcription is extremely error-prone and it is during this step that mutations may occur. Such mutations may cause drug resistance.
RNA replication
RNA replication is the copying of one RNA to another. Many viruses replicate this way. The enzymes that copy RNA to new RNA, called RNA-dependent RNA polymerases, are also found in many eukaryotes where they are involved in RNA silencing.[4] RNA editing, in which an RNA sequence is altered by a complex of proteins and a “guide RNA”, could also be considered an RNA-to-RNA transfer.
Direct translation from DNA to protein
Direct translation from DNA to protein has been demonstrated in a cell-free system (i.e. in a test tube), using extracts from E. coli that contained ribosomes, but not intact cells. These cell fragments could express proteins from foreign DNA templates, and neomycin was found to enhance this effect.[5][6]
Transfers of information not explicitly covered in the theory
Posttranslational modification
Protein amino acid sequence can be edited after translation by various enzymes. This is a case of protein affecting protein sequence, not explicitly covered by the central dogma.
Inteins
An intein is a “parasitic” segment of a protein that is able to excise itself from the chain of amino acids as they emerge from the ribosome and rejoin the remaining portions with a peptide bond. This is a case of a protein affecting its own primary sequence encoded originally by the DNA of a gene. Additionally, most inteins contain a homing endonuclease or HEG domain which is capable of finding a copy of the parent gene not containing the intein nucleotide sequence. On contact with the intein-free copy, the HEG domain initiates the DNA double-stranded break repair mechanism. This process causes the intein sequence to be copied from the original source gene to the intein-free gene. This is an example of protein directly editing DNA sequence, as well as increasing the sequence’s heritable propagation.
Methylation
Variation in methylation states of DNA can alter gene expression levels significantly. Methylation variation usually occurs through the action of DNA methylases. When the change is heritable, it is consideredepigenetic. When the change in information status is not heritable, it would be a somatic epitype. The effective information content has been changed by means of the actions of a protein or proteins on DNA, but the primary DNA sequence is not altered.
DNA methylation
DNA methylation is a biochemical process that is important for normal development in higher organisms. It involves the addition of a methyl group to the5 position of the cytosine pyrimidine ring or the number 6 nitrogen of the adenine purine ring (cytosine and adenine are two of the four bases of DNA). This modification can be inherited through cell division.
DNA methylation is a crucial part of normal organismal development and cellular differentiation in higher organisms. DNA methylation stably alters the gene expression pattern in cells such that cells can “remember where they have been” or decrease gene expression; for example, cells programmed to be pancreatic islets during embryonic development remain pancreatic islets throughout the life of the organism without continuing signals telling them that they need to remain islets. DNA methylation is typically removed during zygote formation and re-established through successive cell divisions during development. However, the latest research shows that hydroxylation of methyl groups occurs rather than complete removal of methyl groups in zygote.[1][2] Some methylation modifications that regulate gene expression are inheritable and are referred to as epigenetic regulation.
In addition, DNA methylation suppresses the expression of viral genes and other deleterious elements that have been incorporated into the genome of the host over time. DNA methylation also forms the basis of chromatin structure, which enables cells to form the myriad characteristics necessary for multicellular life from a single immutable sequence of DNA. DNA methylation also plays a crucial role in the development of nearly all types of cancer.[3]
DNA methylation at the 5 position of cytosine has the specific effect of reducing gene expression and has been found in every vertebrate examined. In adult somatic tissues, DNA methylation typically occurs in a CpG dinucleotide context; non-CpG methylation is prevalent in embryonic stem cells.[4][5][6
DNA methylation is essential for normal development and is associated with a number of key processes including genomic imprinting, X-chromosome inactivation, suppression of repetitive elements, and carcinogenesis.In mammals
Between 60% and 90% of all CpGs are methylated in mammals.[7][8] Methylated C residues spontaneously deaminate to form T residues over evolutionary time; hence CpG dinucleotides steadily mutate to TpG dinucleotides, which is evidenced by the under-representation of CpG dinucleotides in the human genome (they occur at only 21% of the expected frequency).[9] (On the other hand, spontaneous deamination of unmethylated C residues gives rise to U residues, a mutation that is quickly recognized and repaired by the cell.)
Unmethylated CpGs are often grouped in clusters called CpG islands, which are present in the 5′ regulatory regions of many genes. In many disease processes, such as cancer, gene promoter CpG islandsacquire abnormal hypermethylation, which results in transcriptional silencing that can be inherited by daughter cells following cell division. Alterations of DNA methylation have been recognized as an important component of cancer development. Hypomethylation, in general, arises earlier and is linked to chromosomal instability and loss of imprinting, whereas hypermethylation is associated with promoters and can arise secondary to gene (oncogene suppressor) silencing, but might be a target for epigenetic therapy.[10]
DNA methylation may affect the transcription of genes in two ways. First, the methylation of DNA itself may physically impede the binding of transcriptional proteins to the gene, and second, and likely more important, methylated DNA may be bound by proteins known as methyl-CpG-binding domain proteins (MBDs). MBD proteins then recruit additional proteins to the locus, such as histone deacetylases and otherchromatin remodeling proteins that can modify histones, thereby forming compact, inactive chromatin, termed heterochromatin. This link between DNA methylation and chromatin structure is very important. In particular, loss of methyl-CpG-binding protein 2 (MeCP2) has been implicated in Rett syndrome; and methyl-CpG-binding domain protein 2 (MBD2) mediates the transcriptional silencing of hypermethylated genes in cancer.
Research has suggested that long-term memory storage in humans may be regulated by DNA methylation.[11][12]
In cancer
DNA methylation is an important regulator of gene transcription and a large body of evidence has demonstrated that genes with high levels of 5-methylcytosine in their promoter region are transcriptionally silent, and that DNA methylation gradually accumulates upon long-term gene silencing. DNA methylation is essential during embryonic development, and in somatic cells, patterns of DNA methylation are generally transmitted to daughter cells with a high fidelity. Aberrant DNA methylation patterns have been associated with a large number of human malignancies and found in two distinct forms: hypermethylation and hypomethylation compared to normal tissue. Hypermethylation typically occurs at CpG islands in the promoter region and is associated with gene inactivation. Global hypomethylation has also been implicated in the development and progression of cancer through different mechanisms.[13]
DNA methyltransferases
In mammalian cells, DNA methylation occurs mainly at the C5 position of CpG dinucleotides and is carried out by two general classes of enzymatic activities – maintenance methylation and de novomethylation.[citation needed]
Maintenance methylation activity is necessary to preserve DNA methylation after every cellular DNA replication cycle. Without the DNA methyltransferase (DNMT), the replication machinery itself would produce daughter strands that are unmethylated and, over time, would lead to passive demethylation. DNMT1 is the proposed maintenance methyltransferase that is responsible for copying DNA methylation patterns to the daughter strands during DNA replication. Mouse models with both copies of DNMT1 deleted are embryonic lethal at approximately day 9, due to the requirement of DNMT1 activity for development in mammalian cells.
It is thought that DNMT3a and DNMT3b are the de novo methyltransferases that set up DNA methylation patterns early in development. DNMT3L is a protein that is homologous to the other DNMT3s but has no catalytic activity. Instead, DNMT3L assists the de novo methyltransferases by increasing their ability to bind to DNA and stimulating their activity. Finally, DNMT2 (TRDMT1) has been identified as a DNA methyltransferase homolog, containing all 10 sequence motifs common to all DNA methyltransferases; however, DNMT2 (TRDMT1) does not methylate DNA but instead methylates cytosine-38 in the anticodon loop of aspartic acid transfer RNA.[14]
Since many tumor suppressor genes are silenced by DNA methylation during carcinogenesis, there have been attempts to re-express these genes by inhibiting the DNMTs. 5-Aza-2′-deoxycytidine (decitabine) is a nucleoside analog that inhibits DNMTs by trapping them in a covalent complex on DNA by preventing the β-elimination step of catalysis, thus resulting in the enzymes’ degradation. However, for decitabine to be active, it must be incorporated into the genome of the cell, which can cause mutations in the daughter cells if the cell does not die. In addition, decitabine is toxic to the bone marrow, which limits the size of its therapeutic window. These pitfalls have led to the development of antisense RNA therapies that target the DNMTs by degrading their mRNAs and preventing their translation. However, it is currently unclear whether targeting DNMT1 alone is sufficient to reactivate tumor suppressor genes silenced by DNA methylation.
Epigenetics
In biology, and specifically genetics, epigenetics is the study of heritable changes in gene expression or cellular phenotype caused by mechanisms other than changes in the underlying DNA sequence – hence the name epi- (Greek: επί– over, above, outer) -genetics. It refers to functionally relevant modifications to the genome that do not involve a change in the nucleotide sequence. Examples of such changes areDNA methylation and histone modification, both of which serve to regulate gene expression without altering the underlying DNA sequence.
These changes may remain through cell divisions for the remainder of the cell’s life and may also last for multiple generations. However, there is no change in the underlying DNA sequence of the organism;[1]instead, non-genetic factors cause the organism’s genes to behave (or “express themselves”) differently.[2]
One example of epigenetic changes in eukaryotic biology is the process of cellular differentiation. During morphogenesis, totipotent stem cells become the various pluripotent cell lines of the embryo, which in turn become fully differentiated cells. In other words, a single fertilized egg cell – the zygote – changes into the many cell types including neurons, muscle cells, epithelium, endothelium of blood vessels, etc. as it continues to divide. It does so by activating some genes while inhibiting others.[3]
In 2011, it was demonstrated that the methylation of mRNA has a critical role in human energy homeostasis. The obesity associated FTO gene is shown to be able to demethylate N6-methyladenosine in RNA. This opened the related field of RNA epigenetics.[4][5]

Etymology and definitions
Epigenetics (as in “epigenetic landscape”) was coined by C. H. Waddington in 1942 as a portmanteau of the words genetics and epigenesis.[6]Epigenesis is an old[7] word that has more recently been used (see preformationism for historical background) to describe the differentiation of cells from their initial totipotent state in embryonic development. When Waddington coined the term the physical nature of genes and their role in heredity was not known; he used it as a conceptual model of how genes might interact with their surroundings to produce a phenotype.
Robin Holliday defined epigenetics as “the study of the mechanisms of temporal and spatial control of gene activity during the development of complex organisms.”[8] Thus epigenetic can be used to describe anything other than DNA sequence that influences the development of an organism.
The modern usage of the word in scientific discourse is more narrow, referring to heritable traits (over rounds of cell division and sometimes transgenerationally) that do not involve changes to the underlying DNA sequence.[9] The Greek prefix epi- in epigenetics implies features that are “on top of” or “in addition to” genetics; thus epigenetic traits exist on top of or in addition to the traditional molecular basis for inheritance.
The similarity of the word to “genetics” has generated many parallel usages. The “epigenome” is a parallel to the word “genome”, and refers to the overall epigenetic state of a cell. The phrase “genetic code” has also been adapted—the “epigenetic code” has been used to describe the set of epigenetic features that create different phenotypes in different cells. Taken to its extreme, the “epigenetic code” could represent the total state of the cell, with the position of each molecule accounted for in an epigenomic map, a diagrammatic representation of the gene expression, DNA methylation and histone modification status of a particular genomic region. More typically, the term is used in reference to systematic efforts to measure specific, relevant forms of epigenetic information such as thehistone code or DNA methylation patterns.
The psychologist Erik Erikson used the term epigenetic in his book Identity: Youth and Crisis (1968). Erikson writes that the epigenetic principle is where “anything that grows has a ground plan, and that out of this ground plan, the parts arise, each part having its time of special ascendancy, until all parts have arisen to form a functioning whole.”[10] That usage, however, is of primarily historical interest.[11]
Molecular basis of epigenetics
Epigenetic changes can modify the activation of certain genes, but not the sequence of DNA. Additionally, the chromatin proteins associated with DNA may be activated or silenced. This is why the differentiated cells in a multi-cellular organism express only the genes that are necessary for their own activity. Epigenetic changes are preserved when cells divide. Most epigenetic changes only occur within the course of one individual organism’s lifetime, but, if gene disactivation occurs in a sperm or egg cell that results in fertilization, then some epigenetic changes can be transferred to the next generation.[12]This raises the question of whether or not epigenetic changes in an organism can alter the basic structure of its DNA (see Evolution, below), a form of Lamarckism.
Specific epigenetic processes include paramutation, bookmarking, imprinting, gene silencing, X chromosome inactivation, position effect, reprogramming, transvection, maternal effects, the progress ofcarcinogenesis, many effects of teratogens, regulation of histone modifications and heterochromatin, and technical limitations affecting parthenogenesis and cloning.
Epigenetic research uses a wide range of molecular biologic techniques to further our understanding of epigenetic phenomena, including chromatin immunoprecipitation (together with its large-scale variantsChIP-on-chip and ChIP-Seq), fluorescent in situ hybridization, methylation-sensitive restriction enzymes, DNA adenine methyltransferase identification (DamID) and bisulfite sequencing. Furthermore, the use ofbioinformatic methods is playing an increasing role (computational epigenetics).
Pingback: Complete List Of Posts - |
Pingback: A Personal Interpretation And Explanation Of Principles Of Biology And Genetics For The Purpose Of Height Increase | Natural Height Growth
Wow way to blatantly plagiarize Wikipedia.