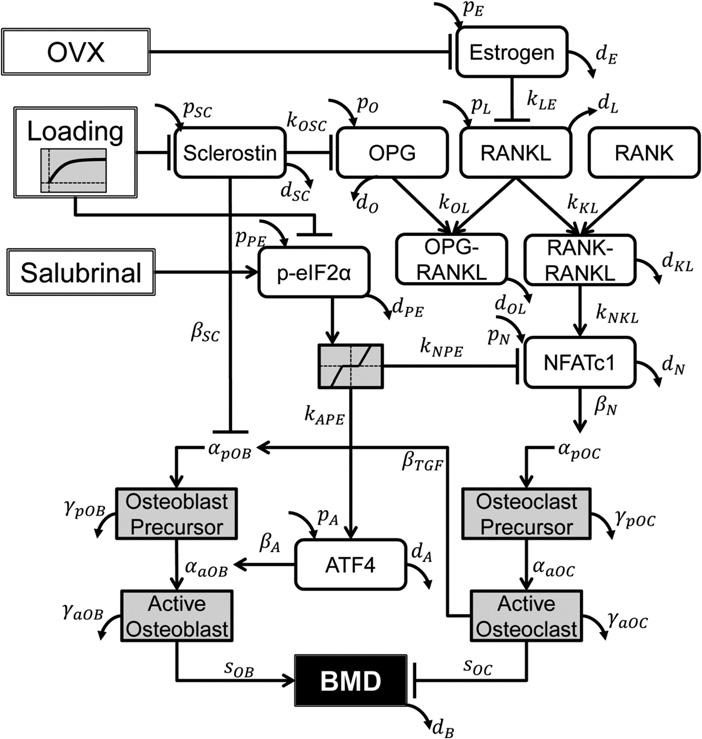
It’s been established that the fingers can grow longer after cessation of development. One primary difference of the fingers and the bones of say the legs is that there’s periosteum covering the articular cartilage of the legs and not of the bones of the fingers. One issue of articular chondrocyte hypertrophy is it’s correlation with osteoarthritis(and).
Chondrocyte hypertrophy in skeletal development, growth, and disease.
” Chondrocyte hypertrophy is an essential contributor to longitudinal bone growth, but recent data suggest that these cells also play fundamental roles in signaling to other skeletal cells, thus coordinating endochondral ossification. On the other hand, ectopic hypertrophy of articular chondrocytes has been implicated in the pathogenesis of osteoarthritis.”
“Chondrocytes first differentiate from mesenchymal precursor cells after these have undergone cellular condensation{we have to be sure we’re covering this step if we want to create new growth plates}. This process, termed chondrogenesis, is characterized by expression of the cartilage master transcription factor Sox9 and induction of its target genes, for example, the classical chondrocyte markers, collagen II, and aggrecan. In a subsequent step, chondrocytes increase their rate of proliferation in a directed pattern along the developing longitudinal axis of the future bone, forming characteristic columns of clonal cells. Under tight control from endogenous and exogenous factors, these cells then withdraw from the cell cycle and start differentiating further. This differentiation step is accompanied by a large increase in cell volume, for example, chondrocyte hypertrophy. The conventional view is that hypertrophic chondrocytes represent the terminal step in this maturation process which culminates in the apoptosis of cells and replacement of hypertrophic cartilage by bone tissue. However, there have been reports about “transdifferentiation” of hypertrophic chondrocytes to osteoblasts”
“Not only is the cell volume increase during hypertrophy a major contributor to longitudinal bone growth, but these cells also act as signaling centers that secrete growth factors, cytokines, and other signaling molecules that can act on other cell types involved in endochondral ossification, such as osteoclasts, osteoblasts, and endothelial cells. Thus, the differentiation to this stage, as well as the behavior of differentiated hypertrophic chondrocytes, are tightly regulated by a multitude of systemic and local factors, including growth hormone, insulin-like growth factors (IGFs), thyroid hormone, parathyroid hormone-related peptide (PTHrP), Indian hedgehog, fibroblast growth factors (FGFs), canonical and noncanonical Wnt signaling, transforming growth factor-β (TGFβ) family members, C-type natriuretic peptide, and others. Within the cell, the classical mediators of these signals, such as β-catenin in canonical Wnt signaling and Smad proteins in TGFβ/bone morphogenetic protein signaling control cartilage growth, along with common kinases (e.g., MAP and PI3K/Akt) and GTPases (e.g., Rho GTPases). In the cell nucleus, Runx and MEF2 transcription factors, along with the histone deacetylase HDAC4, have been recognized as key regulators of chondrocyte hypertrophy.”
“mice lacking both FoxA2 and FoxA3 activity in cartilage display postnatal dwarfism, a result of severely impaired chondrocyte hypertrophy and mineralization shown by markedly attenuated expression of hypertrophic markers, such as collagen X, MMP13, and alkaline phosphatase”
“induction of chondrogenesis in chicken limb-bud mesenchymal micromass cultures results in increased FoxA1 and FoxA2 expression. Electrophoretic mobility shift assays show FoxA2 to bind to conserved binding sites within the chicken collagen X enhancer. Interestingly, exogenous FoxA2 can activate expression of a Collagen X-luciferase reporter gene in both chondrocytes and fibroblasts”
“HIF-2α contributes to physiological endochondral ossification by controlling chondrocyte hypertrophy, cartilage degradation, and vascularization. In particular, HIF-2α was identified as the most potent activator of the COL10A1 promoter in vitro. The expression of HIF-2α’s encoding gene, Epas1, also increased in parallel to that of Col10a1, Mmp13, and Vegf-α during chondrocyte differentiation”
“ectopic expression of Epas1 enhances cytokine-induced expression of catabolic genes necessary for cartilage breakdown. Similarly, both in vivo and in vitro models of Epas1 deficiency show protection against OA through suppression of cartilage degradation and catabolic and hypertrophic gene expression”
” HIF-2α [is a] functional inducer of the CEBPB promoter. As C/EBPβ is a transcription factor crucial for the transition from proliferation to hypertrophy in chondrocytes ”
“HDAC4 (histone deacetylase 4), which inhibits hypertrophy by suppressing the activity of Runx2 and MEF2C”
“kinase SIK3 (salt-inducible kinase 3) is essential to locate HDAC4 in the cytoplasm (and thereby relieve the inhibitory activity of HDAC4 on MEF2C and possibly Runx2). Correspondingly, chondrocytes from Sik3 KO mice show increased nuclear staining for HDAC4, resulting in delayed hypertrophy and dwarfism”
” Hdac3 KO mice showed accelerated chondrocyte hypertrophy but smaller cell size, which the authors attribute to increased expression of the phosphatase leucine-rich repeat phosphatase 1 (Phlpp1) in HDAC3-deficient chondrocytes. Increased Phlpp1 levels then suppress Akt signaling. Since the PI3K-Akt pathway is a major positive regulator of chondrocyte hypertrophy, these data suggest a direct pathway from epigenetic regulation of gene expression to cell size increase”
“Three different phases of hypertrophic cell enlargement, the first characterized by simultaneous increase in cell volume and dry mass, the second by preferential cell swelling without corresponding dry mass production, and the third again by proportional increases in cell volume and dry mass. Differences between slow and fast growing bones were due to changes in phase II and particularly phase III”
“mice deficient for the key collagenase in OA, MMP13, still show chondrocyte hypertrophy in response to OA-inducing joint surgery, but are protected from cartilage degeneration“<-So it’s possible to grow taller via articular cartilage growth without cartilage degeneration.
We know that cartilage hypertrophy plays a very large role in how much bones grow longer. There are supplements that could stimulate chondrocyte hypertrophy but inhibit cartilage degradation. So this sort of method would be possible to test with suppelements only and not need mechanical methods. Cartilage degradation is vital though for longitudinal bone growth via growth plates so this would only work via adults past the developmental stage.
The chondrocytic journey in endochondral bone growth and skeletal dysplasia.
“Extracellular signals, including bone morphogenetic proteins, wingless-related MMTV integration site (WNT), fibroblast growth factor, Indian hedgehog, and parathyroid hormone-related peptide, are all indispensable for growth plate chondrocytes to align and organize into the appropriate columnar architecture and controls their maturation and transition to hypertrophy. Chondrocyte hypertrophy, marked by dramatic volume increase in phases, is controlled by transcription factors SOX9, Runt-related transcription factor, and FOXA2.”
“In vertebrates, the endochondral bones of the axial and appendicular skeleton develop from mesenchymal progenitors that form condensations of bipotential osteo-chondroprogenitors in the approximate shape of the future skeletal elements. These osteo-chondroprogenitors undergo lineage restriction toward either chondrocytes or osteoblasts. The primordial cartilage continues growing through recruitment of more mesenchymal progenitors and proliferation of chondrocytes. Once committed to the chondrocytic fate, the chondroprogenitors further differentiate into chondrocytes which proliferate, mature, exit the cell cycle, and undergo hypertrophy, forming an avascular cartilaginous template surrounded by a perichondrium. Mature hypertrophic chondrocytes secrete matrix vesicles which mediate cartilage calcification. At the same time, adjacent perichondrial cells differentiate into osteoblasts and secrete bone matrix, forming a bone collar surrounding the hypertrophic chondrocytes, then start to organize a periosteum and later become the shaft (diaphysis) of the bone. The primary ossification center begins to develop, with blood vessels penetrating the periosteum and gaining access to the calcified cartilage, bringing in osteoclasts/chondroclasts to degrade the cartilage. Subsequently, the osteoblasts lay down bone matrix to form trabecular bone. Gradually, the chondro-osseous junction forms, dividing the cartilage template into two ends, called epiphyses. These progressive steps of chondrocyte differentiation lead to the formation of a specialized structure called the growth plate ”
“The round chondrocytes of the RZ serve as a pool of progenitor-like cells for subsequent proliferation and differentiation. In the PZ, the round cells divide, become flattened, and organize into columns in the direction of longitudinal growth while proliferating. In the PHZ, the chondrocytes exit the cell cycle and initiate hypertrophic differentiation, characterized by the expression of Ihh (encoding the paracrine factor Indian hedgehog) and Col10a1 (encoding collagen type X). In the HZ, the size of the chondrocytes increases dramatically in the upper hypertrophic zone, and then these hypertrophic chondrocytes terminally differentiate in the lower hypertrophic zone, where the cartilage becomes calcified and is replaced with bone. The fate of hypertrophic chondrocytes at the chondro-osseous junction is controversial, with both cell death and survival being indicated”
Absence of Sox9, BMP2, BMP4, Smad4, Hif1a, and hoxd13 in MSCs all result in reduced height. In proliferating chondrocytes loss of IHH+Gli3, Wnt5a, Wnt5b, Smad1, Smad5, Col2a1, and Acan result in reduced height. In hypertrophic chondrocytes loss of Runx2 and Col10a1 results in reduced height.
“after cell aggregation and cluster formation, BMP signaling promotes cell–cell association through compaction of the aggregate.”
” the growth plates of different bones within the same animal grow at different rates. In part, the difference may be due to the differential duration of the G1 phase in proliferating chondrocytes, suggesting that cell cycle genes regulating G1 progression are of special importance in regulating endochondral bone growth. Progression through the different phases of the cell cycle is controlled by cyclin-dependent kinases (CDKs). The activities of CDKs are in turn regulated in many ways: (1) through the concentrations of their partner proteins, the cyclins; (2) through the concentrations of inhibitory proteins of the CIP/KIP and INK families (CDK inhibitors or CKIs); and (3) through inhibitory and stimulatory phosphorylation of various CDK residues. The principal targets of the CDKs are the pocket proteins RB, RBL1 (also known as p107), and RBL2 (also known as p130). Hypophosphorylated pocket proteins form complexes with transcription factors of the E2F family. Active CDKs phosphorylate the pocket proteins, causing them to dissociate from E2F, freeing the latter to activate target genes involved in cell-cycle progression and DNA replication. Cyclins D1, D2, and D3 are implicated in chondrocyte proliferation. The Cyclin D1 gene is a target of several paracrine factors, including parathyroid hormone-related peptide (PTHrP), IHH, and WNT5b. Mice lacking only cyclin D1 are viable but small, with a smaller PZ in the growth plate. Mouse embryos lacking all three cyclin Ds survive until about E13.5—15.5, suggesting that redundancy exists between the cyclin Ds in proliferation, and that they are regulated by different factors. RB, p107 and p130, regulate cell cycle exit and differentiation of growth plate chondrocytes. The CKI p57Kip2 is strongly expressed in terminally differentiated cells and can mediate cell cycle exit. Mice lacking this gene have short limbs and display endochondral ossification defects, as illustrated by delayed cell cycle exit and incomplete differentiation of hypertrophic chondrocytes. PTHrP and insulin-like growth factor-2 (IGF2) are reported to suppress p57Kip2 gene expression to mediate proliferation. Both IGF2 and CDKN1C (p57Kip2) are located in an imprinted locus and associated with Beckwith-Wiedemann syndrome, an overgrowth syndrome in human.”
“IHH promotes the differentiation of round proliferative chondrocytes in the resting zone into flat column-forming chondrocytes in a Gli3-dependent manner in mice”
“While IHH promotes proliferation, PTHrP delays exit from the cell cycle and initiation of hypertrophic differentiation. PTHrP represses the expression of the cell cycle inhibitor p57Kip2, and promotes the expression of the transcriptional coregulator ZFP521 and cyclin D1 to maintain chondrocyte proliferation”
“PP2A, when activated by PTHrP, dephosphorylates HDAC4, which is then translocated into the nucleus, where it inhibits the function of RUNX2 and MEF2C, and thus inhibits chondrocyte hypertrophy. Conversely, SIK3 can bind to HDAC4 and anchor it in the cytoplasm, leaving MEF2C and RUNX2 free to activate downstream targets to promote chondrocyte hypertrophy”
“Borderline chondrocytes adjacent to the bone collar may differentiate into osteoblasts{It is good for our purposes if chondrocytes transdifferentiate into osteoblasts because that means that they retain some of the chondrocyte genetic coding}. In chick explant culture, surviving hypertrophic chondrocytes undergo asymmetric cell division: one behaves as an osteoblast and may undergo further division while the other undergoes programmed cell death, leaving cell debris in the lacuna. The newly formed osteoblasts in the cartilage lacuna will be released into the chondro-osseous junction and contribute to endochondral bone formation . Some of the hypertrophic chondrocytes look darker under electron microscopy. They are referred to as dark chondrocytes and are proposed to undergo programmed cell death inside the cartilage lacuna”
“[Hypertrophic chondrocytes] have been shown to undergo osteoblastic differentiation in situ when Sox9 is eliminated from prehypertrophic chondrocytes, displaying up-regulation of Runx2, Osx, Alpl, and Col1a1”
“In vitro, isolated chick hypertrophic chondrocytes can differentiate into osteoblasts in the presence of retinoic acid”
“[H1fa] induces collagen prolyl 4-hydroxylase expression in chondrocytes, which is necessary for generating 4-hydroxyprolines for the stability of newly synthesized collagen molecules”
What about avoiding dedifferentiation?
Spontaneous differentiating primary chondrocytic tissue culture: a model for endochondral ossification.
“Primary cartilage-derived cell cultures tend to undergo dedifferentiation, acquire fibroblastic features, and lose most of the characteristics of mature chondrocytes{We want the opposite for it to undergo endochondral ossification, this dedifferentiation could be due to lack of mechanical stimuli}. This phenomenon is due mainly to the close matrix-cell interrelationship typical of cartilage tissue, which is vital for the preservation of the cartilaginous features. In this study we present a model for spontaneous redifferentiation of primary chondrocytic culture. Mandibular condyles excised from 3-day-old mice, thoroughly cleaned of all soft tissue, were digested with 0.1% collagenase. These mandibular condyle-derived chondrocytes (MCDC) were cultured under chondrogenesis-supporting conditions; that is, 5 x 10(5) cells/mL were incubated in Dulbecco’s modified Eagle medium supplemented with 100 microg/mL ascorbic acid, 1 mmol/L calcium chloride, 10 mmol/L beta-glycerophosphate, 10% fetal calf serum, and antibiotics. Development and growth rates of these cartilage-derived cultures were determined by following morphological and functional changes. MCDC proliferated intensively during the first 24-48 h following plating, showing fibroblast-like (long spindle-shaped) morphology and producing mainly type I collagen. The proliferation rate gradually declined, and the cells developed polygonal shapes and started to produce type II collagen. In the 10-14-day-old cultures, cells began to aggregate in cartilaginous nodules and exhibited positive staining for acidic Alcian blue, type X collagen, and von Kossa. Expression of core-binding factor alpha(1) increased between 3 and 5 days and declined gradually thereafter. The condylar-derived tissue culture presented here depicts a spontaneous redifferentiation chondrocytic tissue culture that exhibits features of mature chondrocytes typically found in skeletal growth centers. The present study offers a model for primary chondrocytic tissue culture, which might serve as a model for in vitro endochondral ossification.”
” chondrocytes of the mandibular condyle and the epiphyseal growth plate (EGP) are similarly regulated under both physiological and pathological conditions. Condylar chondrocytes express receptors for growth hormone, IGF-1, and parathyroid hormone and react similarly to the EGP chondocytes in type I diabetes and metabolic acidosis.”